
The Wrong Tool Problem in Genomic AI
A leaderboard tells you which model wins. It can't tell you that you picked the wrong kind of model.


Every Sunday I pick one paper or release that's worth your time, break it apart, and tell you why it matters. No hype. No summaries of summaries. Just the idea, explained.
A researcher walks over with a sequence on her screen. She has a stretch of DNA, a promoter, the switch that turns a gene on, and one variant sitting inside it that she suspects changes how loudly that gene gets expressed. She wants to know if the variant matters. So she asks the question everyone asks now: which AI model should I use? BOLT-LMM or DNABERT?
It sounds like a ranking question. Pick the better of two tools. It is not a ranking question. It is a category error, and it's one I've watched smart teams make without noticing.
BOLT-LMM and DNABERT do not belong in the same column. One is a statistical method built to scan a whole population and find which genetic differences track with a trait across thousands of people. The other reads a single stretch of DNA and turns it into a numeric representation a computer can work with. Asking which is better for a promoter variant is like asking whether a microscope or a telephone is better for measuring temperature. The answer is neither, and the question itself is the problem.
The hardest mistake in genomic AI isn't picking the second-best model. It's picking the wrong kind of model and not knowing it.
The phrase that hides nine different things
"Genomic AI model" sounds like one category. It is at least nine.
There are DNA language models, which read raw sequence and learn its grammar the way a text model learns the grammar of English. There are sequence-to-function predictors, which take a piece of DNA and predict what it does in a cell, how strongly it drives expression, whether a region is open or closed. There are variant-effect predictors, which score how much a specific mutation changes that function. There are statistical-genetics engines, which never look at sequence grammar at all and instead test, across a population, which genotypes associate with which traits. There are single-cell models that work on tables of gene activity per cell, generative models that design new sequences, and a few more.
Every one of these gets called a "genomic AI model." Every one of these shows up in benchmark papers ranked against the others. And every one of these answers a different question.
The researcher with the promoter variant needs a variant-effect predictor or a sequence-to-function model, something that reads her one sequence and tells her what the change does. BOLT-LMM, the statistical-genetics engine, is built for a completely different shape of problem: thousands of people, their genotypes, their traits, and the search for associations across the cohort. It has no way to read the grammar of her single promoter. DNABERT, the DNA language model, reads grammar beautifully but doesn't produce a calibrated answer to "does this variant matter" without more machinery bolted on top.
The two tools the researcher was choosing between were both wrong. The benchmark she'd consult to choose between them can't tell her that.
Why the leaderboard can't catch this
Here is the uncomfortable part. The benchmarks the whole field relies on are structured to make this error invisible.
A leaderboard ranks models within a class. It lines up five DNA language models and tells you which embeds sequence best. It lines up four variant-effect predictors and tells you which scores variants most accurately. That is useful, and the people building those benchmarks are doing careful work. But a leaderboard assumes you already chose the right column. It answers "which is best?" and stays silent on the question that comes before it: best at what, and is that even what I need?
The wrong-class error happens upstream of every benchmark. By the time you're reading a leaderboard, the category decision is already made, usually without anyone noticing a decision was made at all. You typed a model name into a search box, found a paper that ranked it highly, and never asked whether the thing being ranked was the right kind of thing for your task.
Nobody walks into this on purpose. The pull comes from how the field names things. Two models share the word "genomic," both have an impressive accuracy number in a recent paper, both have a HuggingFace page and a star count, and the surface presentation makes them look like rival products on the same shelf. A leaderboard reinforces the illusion by placing them in the same table. Nothing in that experience signals "these answer different questions." The signal you'd need is a layer that sits before the ranking and sorts tools by what they're for, and that layer mostly doesn't exist.
I'd put this error ahead of picking the second-best model in the right class, and it's far more costly. Pick the second-best variant-effect predictor and you lose a few points of accuracy on a task that was at least the right task. Pick a statistical-genetics engine for a single-sequence question and you don't lose accuracy, you get an answer that means nothing, dressed up to look like it means something. The numbers come back formatted, plotted, ready to drop into a slide, and there's nothing on the surface that tells you the foundation was wrong. Weeks of analysis can ride on top of a category mistake made in the first thirty seconds, and the failure surfaces late, if it surfaces at all.
Classify first, rank second
The fix is almost embarrassingly simple to state and surprisingly hard to do without help: classify the computational object before you rank it.
Before you ask which model is best, ask what kind of object your task actually needs. Do you have one sequence and want a functional readout? You need a sequence-to-function or variant-effect model. Do you have a cohort of people and want to find trait associations? You need a statistical-genetics engine. Do you have a table of cells and want to label cell types? A single-cell model. The class is determined by the shape of your data and the shape of your question, not by which model has the most citations this month.
Get the class right and the leaderboard becomes useful again, because now you're ranking within the right column. Get the class wrong and the leaderboard is worse than useless, because it gives you a confident ranking of tools that can't do your job.
The reason this is hard in practice is that the class isn't printed on the box. A model's documentation tells you its architecture and its scores; it rarely says, in plain terms, "this is for cohort association, not single-sequence scoring." You have to infer the class from the shape of the inputs it takes and the outputs it produces, and that inference is exactly the expertise a researcher new to a method doesn't have yet. The whole point of a triage layer is to do that inference for you and to fail loudly when the tool you named is the wrong kind of thing.
What this looks like as a tool
I built a small open thing to make this concrete, because it's easier to argue for "classify first" when you can watch it fire.
It's called ModelMap, and it's a decision layer rather than a leaderboard. You describe your task in plain terms, what you have and what you want, and it triages the class first. When the tool you named doesn't match the class your task needs, it returns a "wrong-tool" verdict: here is the class your task needs, here is the class this tool actually belongs to, and here is why they don't match. Ask it whether DNABERT-2 is right for a population GWAS and it routes you to the statistical-genetics column and flags DNABERT-2 as the wrong class, with the real function of each spelled out.
Underneath are nine method classes and around thirty model cards, with deterministic rules handling the clear-cut category calls and a grounded language-model layer translating free-text questions into the controlled vocabulary. It's a research-use proof of concept, source-available, and you bring your own model key so there's nothing to abuse. I built it because I couldn't find anything that does. The closest existing tool, OmniGenBench, is a leaderboard, excellent at ranking within a class and silent on which class you need.
A leaderboard answers "which is best." The question that wrecks more projects is "best at what, and is that what I need."
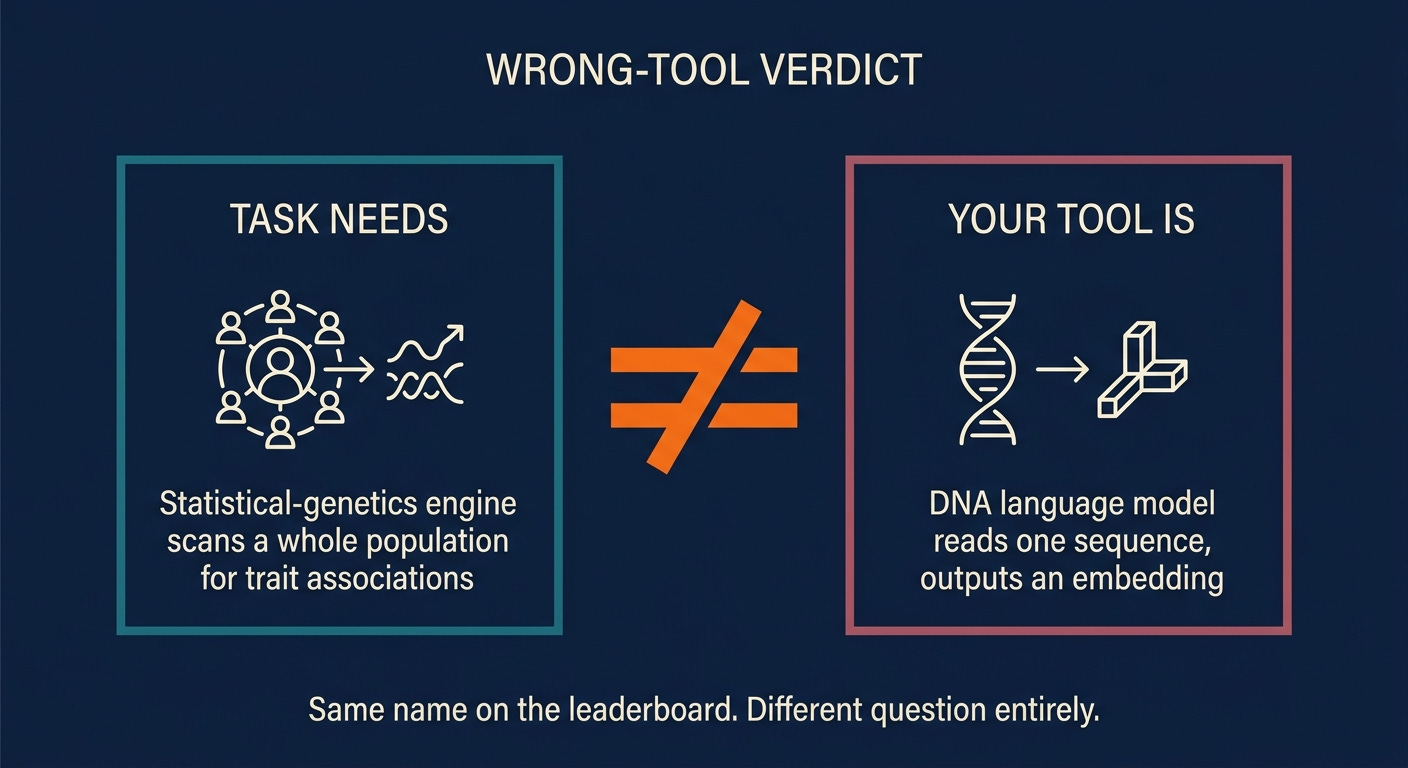
ModelMap's whole point is that one verdict card. It's the moment it tells you the two things you were comparing were never comparable, and points you back one step to the decision you skipped.
If you build tools like this, the harder question is how you let a language model sit in front of that verdict without letting it invent a class or a license. I wrote that part up for the people who'd build it, over on AIXplore: Build an LLM Triage Layer That Can't Freelance.
The question to carry
You don't need ModelMap, and you don't need to remember the nine classes. You need one habit.
The next time someone on your team asks which genomic AI model to use, or which large model, or which forecasting method, or which anything, resist the pull to answer the ranking question they asked. Ask the one underneath it first. What kind of object does this task actually need? Are we even in the right column?
Most of the time the ranking question is the easy part, and the field has built good tools for it. The category question is the one that decides whether the answer means anything, and almost nothing in the standard toolkit asks it for you. That's the gap worth closing, in genomics and well beyond it.
Sunday Deep Dive is a weekly series on Run Data Run. Every Sunday I pick one paper, release, or technique worth understanding, break it apart, and tell you what it means for your work. Free every Sunday, no paywall. If it was useful, the easiest way to support it is to subscribe and forward it to one person on your team who'd want it. If it wasn't, tell me why. I'll make it better.